DNA synthesis and sequencing technology is advancing rapidly, allowing for the design of high-throughput experiments which were previously hindered by technological constraints. Massively Parallel Reporter Assay experiments (MPRAs) is a novel method, that uses the technological advances and the reduction in the associated cost, to study the regulatory activities of tens or hundreds of thousands of DNA oligonucleotides. The oligonucleotides are synthesized using microarrays, each of which contains a uniquely identifiable barcode. Next, the oligonucleotides are amplified, put into vectors with a reporter gene and transfected into cells. By measuring the expression levels of the reporter gene, the regulatory properties of the corresponding sequence can be inferred. Therefore, MPRAs and other similar high-throughput methods, enable the systematic examination of regulatory synthetic sequences to identify rules that govern regulation of gene expression or to examine the type of regulatory role of specific elements, or motif positioning.
For instance, some questions that can be explored using synthetic sequences are:
- The regulatory effect of the positioning of single or combinations of Transcription Factor Binding Sites (TFBSs) such as their relative distance from one another, or their distance from transcription-start sites.
- The type of regulatory effect of a specific element i.e. repressor or activator and the magnitude of its regulatory effect.
- The role of homotypic and heterotypic clusters within regulatory elements and potential synergistic effects.
- The influence of the sequence context and flanking sites at the vicinity TFBSs.
Additionally, MPRAs allow for the effect of regulatory Single Nucleotide Polymorphisms, small insertions or small deletions regarding gene expression. By designing oligonucleotide sequences that differ specifically at a SNP, or contain a small insertions or a small deletions the regulatory role of variants found in the human genome can be examined.
Overall, MPRAs and associated technologies are very powerful, but currently systematically designing these type of experiments remains very challenging. We provide MPRAnator, a novel tool for the design of MPRA experiments to study motifs or polymorphisms in a systematic manner.MPRAs ( Motifs ) query page
This page allows the user to synthesize oligonucleotides for MPRAs experiments to investigate the rules that govern transcription factor occupancy. A set of variables provides fine control of how motifs are placed into the sequences. The user is able to specify the locations to substitute the motifs, insert restriction sites , adapter sites and barcodes.
Parameters :
Enter FASTA sequences: The Fasta sequences as text.Upload FASTA sequences: The Fasta sequences as a text file.Enter your motifs: The motifs in FASTA format.Reverse complement sequence: The option to reverse complement the sequences before motif substitution.Minimum Spacing: The minimum spacing between the motifs.Maximum Spacing: The maximum spacing between the motifs.Distance from left edge: This is the minimum distance from left-most nucleotide of the motif to the left edge of the sequence.Distance from right edge: This is the minimum distance from right-most nucleotide of the motif to the right edge of the sequence.Interval of substitution of motifs: This specifies the interval length to insert the motifsBarcode length: The size of the barcode specifiedMinimum barcode GC content (%): The minimum amount (in percent) of GC content in each of the barcodesMaximum barcode GC content (%): The maximum amount (in percent) of GC content in each of the barcodesBarcode edit distance: The Levenshtein distance between each barcode. The default is 2.Number of barcodes per sequence: This specifies the number of barcodes inserted per sequence (replicates).Restriction sites: The ability to add upto 2 restriction sites into the final product.Adapter sites: The ability to add upto 2 adapter sites into the final product.Ordering: Allows the user to order the constituent parts therefore providing flexibility in the design of the experiment.
MPRAs ( Motifs ) result page
The result page displays the synthesized
oligonucleotides in the FASTA format. The user is able to download ( plain text file ) the generated oligonucleotides.
The description line (header) has information about the
options chosen by the user during submission. A header is composed of
one or more DESCRIPTORs and each DESCRIPTOR is composed of a LABEL and
INFO. The descriptors are delimited by a |, i.e. a "pipe".
Image below is an excerpt of the Results page ( showing 8 nucleotides ). The nucleotides in red are the substituted motifs.

The format of the FASTA header for each sequence is shown below.
Note that the order of the DESCRIPTOR in the header is insignificant.
>
<LABEL> - <INFO> |
<LABEL> - <INFO> |
<LABEL> - <INFO> |
...
The LABEL is one of the options as shown below and INFO is either
a number or a word which describes the LABEL in more detail.
LABEL types:
<MOTIF>: A particular motif inserted by the user.BARCODE: The variant of the same sequenceRESTRICTION: This is the restriction site put by the userADAPTER: The presence of the adapter sequence of the specified numberDUPLICATE_RESTRICTION_SITES: The restriction site which has multiple copies present.
Example of a header:
> ATGTG - 53|AAAAA-61|RESTRICTION - 1|RESTRICTION - 2
There are 4 DESCRIPTORs.
ATGTG - 53is the motif starting at position 54 in the background sequence.AAAAA - 61is the motif starting at position 61 in the background sequence.RESTRICTION - 1signifies the presence of the restriction site 1 in the final sequence.RESTRICTION - 2signifies the presence of the restriction site 2 in the final sequence.
MPRAs ( SNPs ) query page
This page allows the user to synthesize oligonucleotides for MPRA experiments to study the effects of SNPs. The user can select to include or exclude combinations of SNPs when designing MPRA experiments.
Parameters :
Enter FASTA sequences: Sequence in Fasta Format. Header contains the location information.Upload FASTA sequences: Sequence in Fasta Format as a text file. Header contains the location information.Enter your SNPs (VCF Format): SNPs in VCF format (as text).Make Snp Combinations?: Create all combinations of SNPs to be substituted to sequences.Barcode length: The size of the barcode specifiedMinimum barcode GC content (%): The minimum amount (in percent) of GC content in each of the barcodesMaximum barcode GC content (%): The maximum amount (in percent) of GC content in each of the barcodesBarcode edit distance: The Levenshtein distance between each barcode. The default is 2.Number of barcodes per sequence: This specifies the number of barcodes inserted per sequence (replicates).Restriction sites: The ability to add upto 2 restriction sites into the final product.Adapter sites: The ability to add upto 2 adapter sites into the final product.Ordering: Allows the user to order the constituent parts therefore providing flexibility in the design of the experiment.
MPRAs ( SNPs ) result page
The result page (plain text view) displays the synthesized oligonucleotides in FASTA format. The description line (header) has information about the options chosen by the user during submission. A header is composed of one or more DESCRIPTORs and each DESCRIPTOR is composed of a LABEL and INFO. The descriptors are delimited by a |, i.e. a "pipe".
>
<LABEL> - <INFO> |
<LABEL> - <INFO> |
<LABEL> - <INFO> |
...
LABEL types:
<SNP>: Name of SNP<NUCLEOTIDE>: REF / ALTBARCODE: This is the unique identifier for the variantsRESTRICTION: This is the restriction site(s) put by the userADAPTER: This is the adapter site(s) put by the userDUPLICATE_RESTRICTION_SITES: Report the restriction site which has multiple copies present in the oligo
Transmutation query page
Transmutation tool allows for the design of relevant controls for MPRA experiments. For instance, the regulatory role of a motif can be destroyed by introducing one or multiple random mutations. This page provides 4 options:- Mutate random positions in the input sequence.
- Scrambling sequences.
- Reverse sequences.
- Complement sequences.
The input will accept all nucleic acid IUPAC letters.
Transmutation result page
Scrambled or/and mutated sequences in FASTA format. The scrambling, reversing and complementation is implemented in the order as shown in the header.Example of a header:
> sequence2|Mutated_nucleotides - 3|Scrambled - No| Reverse - No | Complemented - Yes
There are 3 DESCRIPTORs.
sequence2This is the header of the sequenceMutated_nucleotides - 3This is the number of randomly chosen mutated nucleotides.Scrambled - NoDenotes whether the sequence was scrambled.Reversed - NoDenotes whether the sequence was reversed.Complemented - NoDenotes whether the sequence was complemented.
PWM-SeqGen query page
- Sequences generated by simulations - This uses the probabilities in the PWM
- All possible sequences generated - This uses the probabilities and removes any low probability k-mer sequences using the threshold
There are 2 ways to use this tool.
PWM Seq-Gen allows for stochastic conversion of Position Weight Matrices (PWMs) into Motif Sequences, using the weights for each nucleotide at each position for the corresponding matrices. The output of PWM Seq-Gen can be inputted into MPRAs Motifs tool, therefore allowing for the design of MPRA experiments using PWMs.
Parameters:
- Generating all possible motifs: This generates all motifs with their probabilities
- Threshold: This is a threshold to remove any sequences with low probabilities
- Enter the Position Weight Matrices: Header starting with either “>”. Matrix should be tab or space separated and containing either integers or decimal numbers. Decimal numbers should be DOT (“.”) separated not COMMA (“,”) separated.
- Enter the number of simulations: Number of times to convert each PWM into a Sequence Motif
- Remove duplicates: Allows users to either keep only unique Sequence Motifs or include Duplicates as well.
PWM-SeqGen result page
The result page displays the generated Motif Sequences in FASTA format. The description line (header) includes the name of the motif and the simulation number.Example of a header:
> MA0056.1 MZF1_1-4 | Simulation number - 4
There are 2 DESCRIPTORs.
MA0056.1 MZF1_1-4This is the header of the simulated motif.Simulation number - 4This is the simulation number of the PWM.
Motif Use Case
Inspired by the results presented by Nguyen et al, we would like to investigate the effects of AP1 (TGACTCA), ELK1 (ACCGGAAGT) and RFX (CGTTGCTAGGCAACG) on gene expression. In the original study, the authors focused on one motif at a time to show that they have a strong impact on the ability of a sequence to act either as a promoter or as an enhancer. It was also shown that the effect of each motif can vary significantly depending on the background.
To explore the impact of different backgrounds, different motif combinations and different spacings, we use the following settings:
First we pick two regions from the mouse genome that do not show any regulatory activity according to the available information from the sources available through the UCSC genome browser. The center of each tile (in mm9 coordinates) is reported in the header.
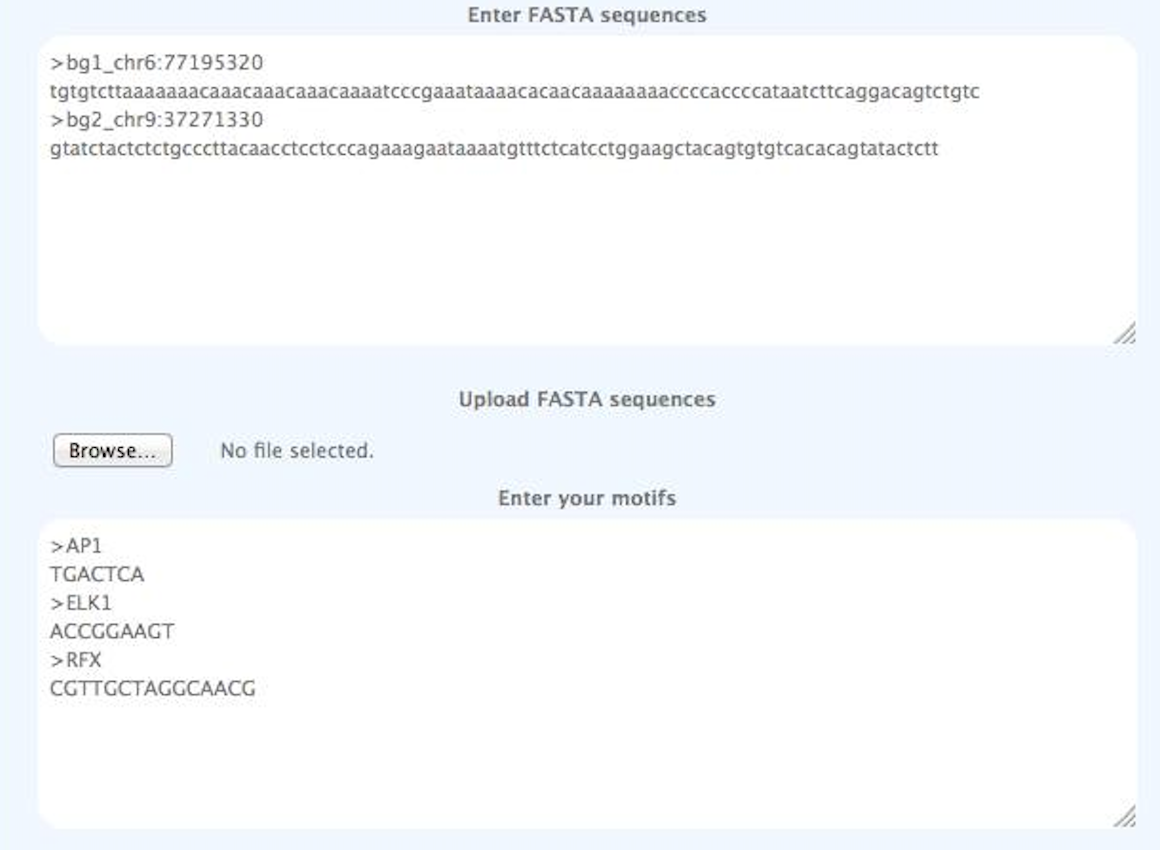
Sequence and Motif examples
>bg1_chr6:77195320
tgtgtcttaaaaaaacaaacaaacaaacaaaatcccgaaataaaacacaacaaaaaaaaccccaccccataatcttcaggacagtctgtc
>bg2_chr9:37271330
gtatctactctctgcccttacaacctcctcccagaaagaataaaatgtttctcatcctggaagctacagtgtgtcacacagtatactctt
The following motifs are used:
>AP1
TGACTCA
>ELK1
ACCGGAAGT
>RFX
CGTTGCTAGGCAACG
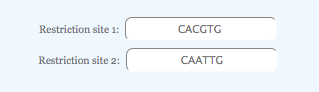
Restriction sites
Here we assume that the total tile length is 117 base pairs. For the experimental design, two restriction sequences are necessary: CACGTG and CAATTG. The restriction sites are rearranged so that they flank the background.
Barcodes
Since this gives us 90 bps to place three motifs and a barcode, we use 15 bps for the barcodes, and to reduce the bias we limit the GC-content of the barcodes to between 40 and 60%. We require an edit distance of 3 and we use 6 different barcodes for each sequence.

Spacing
Due to the relatively long tiles, we allow a minimum of 15 bps to each edge. Finally, to space the motifs we set the minimum and maximum spacings to 6 and 24, respectively, and the interval of substitution is set to 12.
This results in a total of 5856 sequences.